This one is almost guaranteed to be confusing. I will do my best. As always, if you have any question, please ask in the comments or an email, and I will try to address it.
I've told you about playing with cells. So what happens after they are transfected? In my case, I transfected them because I wanted them to express a certain protein (my protein of interest is called BORIS). So now, I want access to that protein. BORIS is a protein that binds to DNA. DNA is inside the nucleus of the cell. If you can reach back with me to high school biology, you might remember that a cell has a membrane that is sort of like skin. It lets certain things in (ions, glucose, etc.) and tries to keep certain things out (viruses for instance). It holds the cell together as a singular unit. Inside the cell membrane is the cytosol. This is basically a fancy way of saying the innards of the cell. This includes the all the structures in the cell, the liquid inside the cell, all the proteins that are chilling in there, and a bunch of other stuff that I have no interest in.
I want the nucleus. The nucleus is sort of like a mini cell within the cell. It has a membrane around its outside also, called the nuclear envelope. Inside, is the nucleus, which by our analogy is like the cytosol. It contains DNA and other goodies. So, how do I get the nucleus, and get rid of the cell membrane and the cytosol? The answer is nuclear extraction.
You have probably read that I do a lot of hurry up and wait. That on any given day, I probably don't do more than 2 hours of actual work. That is the antithesis of nuclear extraction. The first time I did a nuclear extraction, it took me about six hours from start to finish. Six hours of actually doing stuff. I have gotten a lot better at it, where my last attempt only took about 4 hours.
First you need to harvest your cells. I have generally done between 15 and 20 plates at a time with my nuclear extractions. I leave most of them in the incubator and harvest only one of my transfected cell types. For example, I am working with mutants of the BORIS protein and I have six that I work with (where one of them is the "wild-type" unmodified protein). I will generally have five plates of any given type of BORIS-protein-expressing-cells. So I take five plates, vacuum off the media (food) from the cells while trying not to actually vacuum up any cells. I then wash the plate gently and vacuum off the wash (again trying to miss the cells). Then, you have to scrape the cells off the plate. This is done with a tool called a cell scraper. It is like a mini squeegee attached to a toothbrush handle. You scrape this along the bottom of the plate, and try to get all the cells to come off and congregate in one place. Then you suck off the cells, and put each plate's worth of cells into a test tube. Repeat this process for all of your plates and you will have harvested about 10^7 cells from each plate.
Next, you stick all those test tubes into a centrifuge, and spin them around at a lazy pace of 300 x the acceleration due to gravity. Hard to believe, but that actually is a lazy pace. The centrifuge can do 14,000 x the acceleration of gravity. The point of this is to pull all the heavy stuff (the cells actually) to the bottom, and leave the liquid separate. Then you try to pipette off the liquid so you are left with just the cells. Then you wash the cells with the same stuff as before, but mixed with some ingredients that will try to prevent the cells from dying/committing suicide (when cells are shocked enough, they will die and send signals to their buddies to kill themselves). Again you stick them in the centrifuge, and again you suck off the fluid, and again wash them. Repeat this previous sentence.
The reason that the centrifuging was at such a slow pace was that we didn't want to rupture the cells. We just wanted to wash them a bit. Now we mix them with hypotonic buffer. The inside of the cell wants to be at equilibrium with the outside of the cell. This is generally done by controlling the amounts of ions that pass through the membrane. This will then change the amount of water in the cell by osmosis. The hypotonic buffer will make the cells swell up.
You let the cells swell for a little while then add detergent. The detergent will break apart the cell membrane. Stick these broken up cells in the centrifuge, and the liquid portion will be the cytosol. Pipette that off and if you are interested in the cytosol you can store it for use later.
I, however, am interested only in the nuclear fraction. I just throw away the cytosol at this point. Now, you add nuclear extraction buffer (creative name, huh?). You shake/mix vigorously for a few seconds. Let it sit for a few minutes. Shake/mix vigorously for a little longer. Let it sit for a few. This should have turned the nuclear envelope inside out (I'm not so clear on how it does this...). Then you centrifuge at 14,000 x gravity. The liquid portion of this is the nuclear fraction, and the pellet (the non-disolved stuff at the bottom) is cell membranes and nuclear envelope and other stuff that we don't want. We pop the nuclear fraction into fresh tubes, and stick it in the -80 C (otherwise known as really frickin' cold) freezer, and we are done.
Friday, July 2, 2010
Tuesday, June 22, 2010
new students
I admit that I often get bored at the lab. Sometimes I have very little to do, or am waiting on a particular thing to finish incubating.
It is 1:15 in the afternoon and I have not yet done science. But, I am a t-shirt and "New Student Kit" richer.
Today was the second in the "Summer Student Seminar Series." Basically, a bunch of summer interns sit in an auditorium and hear about new/interesting research. The speaker who was supposed to come in today cancelled, but they had a different researcher step in. He was actually quite interesting.
Last week, at the seminar, I only nodded-off three times. Today, there was zero nodding-off. The speaker knew how to engage the audience (students, mostly) and he asked questions. Correct answers warranted a Snickers bar. I had three correct answers, but only needed one Snickers. He spoke to us about Lupus, and specifically how it seems that he accidentally infected mice with it. This is currently (as I write this) giving rise to a new model of Lupus. As I said before, it was quite interesting.
At the end of the seminar, there were student intern t-shirts to be had. In addition, New England BioLabs prepared welcome kits for us. It contained a lot of crap mostly. There were posters, catalogs, and lots of other things that I have not yet gone through, but most intriguing there were samples of reagents. In fact, they basically sent all the things necessary to do a PCR or Polymerase Chain Reaction (read about that in earlier posts). Also, they gave me a retractable super-fine tip Sharpie permanent marker. I always like goody bags.
Now that the seminar is over, I am waiting for the next 41 minutes, 46 seconds for something to finish incubating.
It is 1:15 in the afternoon and I have not yet done science. But, I am a t-shirt and "New Student Kit" richer.
Today was the second in the "Summer Student Seminar Series." Basically, a bunch of summer interns sit in an auditorium and hear about new/interesting research. The speaker who was supposed to come in today cancelled, but they had a different researcher step in. He was actually quite interesting.
Last week, at the seminar, I only nodded-off three times. Today, there was zero nodding-off. The speaker knew how to engage the audience (students, mostly) and he asked questions. Correct answers warranted a Snickers bar. I had three correct answers, but only needed one Snickers. He spoke to us about Lupus, and specifically how it seems that he accidentally infected mice with it. This is currently (as I write this) giving rise to a new model of Lupus. As I said before, it was quite interesting.
At the end of the seminar, there were student intern t-shirts to be had. In addition, New England BioLabs prepared welcome kits for us. It contained a lot of crap mostly. There were posters, catalogs, and lots of other things that I have not yet gone through, but most intriguing there were samples of reagents. In fact, they basically sent all the things necessary to do a PCR or Polymerase Chain Reaction (read about that in earlier posts). Also, they gave me a retractable super-fine tip Sharpie permanent marker. I always like goody bags.
Now that the seminar is over, I am waiting for the next 41 minutes, 46 seconds for something to finish incubating.
Thursday, May 27, 2010
Cells
You know those little guys that we all learned in high school biology that are the building blocks of life? Yea, they're important. I do a lot of work with cells, and I want to talk about some of those procedures.
To begin with, the type of cells that I work with are Human Embryonic Kidney 293T cells. Known as HEKs or 293Ts. A cell line is a group of immortalised cells. You can order them from a company, and you know what you are getting. HEK 293s were originally from a "healthy aborted fetus" in the 1970s. The number 293 comes from Frank Graham (the guy who immortalised this cell line), who numbered each of his experiments. Thus, HEK 293s came from his 293rd experiment.
HEK 293s are not just plain human cells. A bit of adenovirus 5 DNA was added and incorporated into the human chromosome. This is what gives them their immortality.
Ours are not just plain 293s, but 293Ts. This means that they are able to be transfected with plasmids easily. More on that later.
The way we work with these cells is very cyclical, so I really could start anywhere in describing the process.
We'll start with a flask filled with cells. The cells are in media (read food). We use DMEM as our media. DMEM is composed of amino acids, salts, glucose, vitamins, iron, and phenol red. Phenol red is very important. It is a dye that changes color based on how acidic it is. Mixed into our DMEM is fetal bovine serum (FBS) and a penicillin/streptomycin mixture. The FBS is what is left of the liquid portion of fetal cow blood after it is allowed to clot. It does not have blood cells in it, is low in antibodies, and has a good amount of growth factors. The pen/strep mixture is antibiotics so that the cells don't get infected.
You pull your flask out of the incubator (37 degrees C and 5% CO2), and you notice that the liquid (media) is a dull orange. Time to change the media! Why? Well, remember that phenol red? It starts out a reddish-pinkish color. When it gets too acidic (cell excrement makes it that way), the cells will die. Imagine living in your own poop. Even if you get nutrients from the air, after a while there is just too much poop, and you suffocate from it. Same sort of thing. The change in color signifies that it is becoming too acidic. These particular cells are called adherents. This means that they stick to a surface. In this case, they stick to a side of the flask (whichever end was down). So you flip the flask over, and all the liquid is on the opposite side from the cells. Now you can use an aspirating pipette (also known as a vacuum) to pull out the spent media. If you just want your cells to grow more, you will add more media. This is known as changing, swapping, or replacing the media.
Let's say that before you swapped the media, you looked at your flask under a microscope. You found that there was some overcrowding going on. This is another leading cause of cell death. If cells do not have space to grow in, they die. Sometimes something peculiar happens and the cells on the edge will start growing up the walls. Sometimes, they don't have good anti-gravity skills and the off-the-wallers become curlers. A film of cells will curl up off the wall and back into the cells. It may look cool, but it definitely means your cells are unhealthy (I know from experience).
How do you deal with cell-overcrowding? Decimation. You suck off the old media, and put in a little bit of trypsin-EDTA. The trypsin will cleave proteins (it's actually one of the digestive enzymes that digests proteins in humans), and the EDTA prevents the cells from clumping. You leave that in the incubator for five minutes, and all your cells will come off the walls/floor of the flask. You then spray them down with media so any clingers are forced into the solution. You pipette up and down to make sure everything is mixed nicely, and usually you pull off 9/10 of the liquid (and thus the cells) in the pipette. Then you vaccuum them. You are left with 1/10 your original cell count (-ish) and you add more media to get back to your original volume. I have found that doing this generally gives you three or four days before your cells are confluent (overcrowding) again.
We have cells. We know how to maintain them. What's the point? In our case, the point is usually transfection. Transfection is introducing foreign DNA for the cell to incorporate in its own genome. You put your cells in solution, and instead of vaccuuming a large portion of them, you then add that portion to a different flask. You then add an appropriate amount of media, and do some heavy mixing. Then you add about 10 ml of this new solution to your cell culture plates. You may know them as petri dishes. Your cells will grow and flourish (hopefully), and reach confluency.
After a couple of days, when it is time to change the media, you can transfect them. You put a bit of the DNA that you hope to express in a mix with media and a special reagent that will make a hole in the cell membrane for the DNA to go through. You add a few drops of this mix to each plate, and the new foreign DNA gets pulled into the cell. If everything worked well, the cell will think, 'Huh! There is DNA outside of the nucleus. Better put that back where it belongs!' The cell adds the new DNA to its own, and then acts as though nothing is different. In our case, the foreign DNA will code for a particular protein.
After another couple of days, the cells had enough time to produce that protein, and you can harvest them. This is done by basically scraping each of the plates, and sucking up the cells that were on there. There are a number of things that can be done after you have harvested the cells, but that is outside the scope of this post.
Now, you are an expert on cell culture! (-ish)
To begin with, the type of cells that I work with are Human Embryonic Kidney 293T cells. Known as HEKs or 293Ts. A cell line is a group of immortalised cells. You can order them from a company, and you know what you are getting. HEK 293s were originally from a "healthy aborted fetus" in the 1970s. The number 293 comes from Frank Graham (the guy who immortalised this cell line), who numbered each of his experiments. Thus, HEK 293s came from his 293rd experiment.
HEK 293s are not just plain human cells. A bit of adenovirus 5 DNA was added and incorporated into the human chromosome. This is what gives them their immortality.
Ours are not just plain 293s, but 293Ts. This means that they are able to be transfected with plasmids easily. More on that later.
The way we work with these cells is very cyclical, so I really could start anywhere in describing the process.
We'll start with a flask filled with cells. The cells are in media (read food). We use DMEM as our media. DMEM is composed of amino acids, salts, glucose, vitamins, iron, and phenol red. Phenol red is very important. It is a dye that changes color based on how acidic it is. Mixed into our DMEM is fetal bovine serum (FBS) and a penicillin/streptomycin mixture. The FBS is what is left of the liquid portion of fetal cow blood after it is allowed to clot. It does not have blood cells in it, is low in antibodies, and has a good amount of growth factors. The pen/strep mixture is antibiotics so that the cells don't get infected.
You pull your flask out of the incubator (37 degrees C and 5% CO2), and you notice that the liquid (media) is a dull orange. Time to change the media! Why? Well, remember that phenol red? It starts out a reddish-pinkish color. When it gets too acidic (cell excrement makes it that way), the cells will die. Imagine living in your own poop. Even if you get nutrients from the air, after a while there is just too much poop, and you suffocate from it. Same sort of thing. The change in color signifies that it is becoming too acidic. These particular cells are called adherents. This means that they stick to a surface. In this case, they stick to a side of the flask (whichever end was down). So you flip the flask over, and all the liquid is on the opposite side from the cells. Now you can use an aspirating pipette (also known as a vacuum) to pull out the spent media. If you just want your cells to grow more, you will add more media. This is known as changing, swapping, or replacing the media.
Let's say that before you swapped the media, you looked at your flask under a microscope. You found that there was some overcrowding going on. This is another leading cause of cell death. If cells do not have space to grow in, they die. Sometimes something peculiar happens and the cells on the edge will start growing up the walls. Sometimes, they don't have good anti-gravity skills and the off-the-wallers become curlers. A film of cells will curl up off the wall and back into the cells. It may look cool, but it definitely means your cells are unhealthy (I know from experience).
How do you deal with cell-overcrowding? Decimation. You suck off the old media, and put in a little bit of trypsin-EDTA. The trypsin will cleave proteins (it's actually one of the digestive enzymes that digests proteins in humans), and the EDTA prevents the cells from clumping. You leave that in the incubator for five minutes, and all your cells will come off the walls/floor of the flask. You then spray them down with media so any clingers are forced into the solution. You pipette up and down to make sure everything is mixed nicely, and usually you pull off 9/10 of the liquid (and thus the cells) in the pipette. Then you vaccuum them. You are left with 1/10 your original cell count (-ish) and you add more media to get back to your original volume. I have found that doing this generally gives you three or four days before your cells are confluent (overcrowding) again.
We have cells. We know how to maintain them. What's the point? In our case, the point is usually transfection. Transfection is introducing foreign DNA for the cell to incorporate in its own genome. You put your cells in solution, and instead of vaccuuming a large portion of them, you then add that portion to a different flask. You then add an appropriate amount of media, and do some heavy mixing. Then you add about 10 ml of this new solution to your cell culture plates. You may know them as petri dishes. Your cells will grow and flourish (hopefully), and reach confluency.
After a couple of days, when it is time to change the media, you can transfect them. You put a bit of the DNA that you hope to express in a mix with media and a special reagent that will make a hole in the cell membrane for the DNA to go through. You add a few drops of this mix to each plate, and the new foreign DNA gets pulled into the cell. If everything worked well, the cell will think, 'Huh! There is DNA outside of the nucleus. Better put that back where it belongs!' The cell adds the new DNA to its own, and then acts as though nothing is different. In our case, the foreign DNA will code for a particular protein.
After another couple of days, the cells had enough time to produce that protein, and you can harvest them. This is done by basically scraping each of the plates, and sucking up the cells that were on there. There are a number of things that can be done after you have harvested the cells, but that is outside the scope of this post.
Now, you are an expert on cell culture! (-ish)
Thursday, May 20, 2010
General Updates
A lot has happened in the last few weeks. To start with, I no longer live with JK. He went away on vacation for about a week, I watched Hershey the dog, and when he came back, I moved out. This was planned, and I gave plenty of headway.
I currently am living in an apartment that's actually in the center of town. It is a little bit less than two miles to get to the base. In fact, I turn right out of my apartment, turn right at the end of the street, and if I keep going straight, I end up on base. Win.
AK finished up school for the year, and came down to live with me. Timing worked out really well there. She finished up her finals and whatnot before the end-end, and I had a meeting to finish up Division II with JM and CJ. I went up to Amherst for my meeting, brought some great beer to celebrate (Three Philosophers. If you haven't tried it, do so). JM and CJ asked me all sorts of questions to gauge the sorts of things I have learned in the last two years. This is, of course, after I had given them a portfolio and retrospective essay. That way, they knew about which things to ask questions.
After the meeting, AK and I drove down to my folks' house. We had some dinner and grabbed a few things that I had not brought down the first time around. In the morning, we drove down to Frederick. And, I, smartypants that I am, went in to work. I had a nuclear extraction to do (I will explain that in more detail in a different post), and it took me about five hours. I had just driven about five hours, and then I worked for five hours. A glutton for punishment.
AK and I went tag saling last weekend. We bought a table, chairs, a microwave, a toaster, and various knick-knacks. Our apartment is starting to look homely.
AK is on the hunt for the elusive "job." She has applied to probably 30 places by now... So far, no dice. But, not all of them have rejected her yet either. If nothing comes, she is planning on volunteering. Probably with the local animal shelter.
I have been falling more and more into a routine. Aside from nuclear extractions that take about five continuous hours, it is mostly hurry up and wait. Right now, I am waiting for a gel electrophoresis (1.5 hours). After that, I do about ten minutes of stuff, and wait another 1.5 hours, etc.
I currently am living in an apartment that's actually in the center of town. It is a little bit less than two miles to get to the base. In fact, I turn right out of my apartment, turn right at the end of the street, and if I keep going straight, I end up on base. Win.
AK finished up school for the year, and came down to live with me. Timing worked out really well there. She finished up her finals and whatnot before the end-end, and I had a meeting to finish up Division II with JM and CJ. I went up to Amherst for my meeting, brought some great beer to celebrate (Three Philosophers. If you haven't tried it, do so). JM and CJ asked me all sorts of questions to gauge the sorts of things I have learned in the last two years. This is, of course, after I had given them a portfolio and retrospective essay. That way, they knew about which things to ask questions.
After the meeting, AK and I drove down to my folks' house. We had some dinner and grabbed a few things that I had not brought down the first time around. In the morning, we drove down to Frederick. And, I, smartypants that I am, went in to work. I had a nuclear extraction to do (I will explain that in more detail in a different post), and it took me about five hours. I had just driven about five hours, and then I worked for five hours. A glutton for punishment.
AK and I went tag saling last weekend. We bought a table, chairs, a microwave, a toaster, and various knick-knacks. Our apartment is starting to look homely.
AK is on the hunt for the elusive "job." She has applied to probably 30 places by now... So far, no dice. But, not all of them have rejected her yet either. If nothing comes, she is planning on volunteering. Probably with the local animal shelter.
I have been falling more and more into a routine. Aside from nuclear extractions that take about five continuous hours, it is mostly hurry up and wait. Right now, I am waiting for a gel electrophoresis (1.5 hours). After that, I do about ten minutes of stuff, and wait another 1.5 hours, etc.
The smallest things
Today, a package came for AB (not an altogether uncommon experience). It was probably about 8x6x5 inches. Inside it was another box, about half the size, with nothing else. Fischer Scientific (the sender) is claiming to go green, but I have yet to see any evidence.
Inside this second box, was... labelling tape. Twelve rolls. This is basically colored masking tape, but a little bit stronger so you can color code/label your experimental items. There was only one roll each of blue and orange, but two rolls of white, yellow, green, pink, and red. I asked AB, and he said that blue and orange are the most popular colors so they sell those separately. No, that does not make any sense to me either.
AB decided he had to put the colors in the right, most useful order. We have a tape dispenser that can accomodate eight rolls of tape, so that is one of each color, plus one of scotch-type transparent tape. AB spent the next five to ten minutes putting them in the "right" order. Previously, I had just used whatever tape was closest or whatever, but now it seemed like there was a correct and incorrect tape to use for different circumstances. I asked AB. He said, "Of course the colors have meaning. Everything has meaning." I made a face that begged for a more complete response, but none was forthcoming.
He had seven rolls on there when he said, "Hmm, but where does the pink go?" He thought about it for a moment, and took a few rolls off, put the pink on then replaced the rest. The final order is transparent, orange, red, pink, yellow, green, blue, white. He glanced up at me and looked really pleased with himself. I had an urge to pat him on the head and say, "Good boy!"
It's the smallest things that make people happy.
Inside this second box, was... labelling tape. Twelve rolls. This is basically colored masking tape, but a little bit stronger so you can color code/label your experimental items. There was only one roll each of blue and orange, but two rolls of white, yellow, green, pink, and red. I asked AB, and he said that blue and orange are the most popular colors so they sell those separately. No, that does not make any sense to me either.
AB decided he had to put the colors in the right, most useful order. We have a tape dispenser that can accomodate eight rolls of tape, so that is one of each color, plus one of scotch-type transparent tape. AB spent the next five to ten minutes putting them in the "right" order. Previously, I had just used whatever tape was closest or whatever, but now it seemed like there was a correct and incorrect tape to use for different circumstances. I asked AB. He said, "Of course the colors have meaning. Everything has meaning." I made a face that begged for a more complete response, but none was forthcoming.
He had seven rolls on there when he said, "Hmm, but where does the pink go?" He thought about it for a moment, and took a few rolls off, put the pink on then replaced the rest. The final order is transparent, orange, red, pink, yellow, green, blue, white. He glanced up at me and looked really pleased with himself. I had an urge to pat him on the head and say, "Good boy!"
It's the smallest things that make people happy.
Wednesday, May 5, 2010
Umm...News?
First, I realize it's been a while since last I updated you on life in the lab. I have been falling into a bit of routine. I now have my very own laboratory notebook, in which I need to detail my experiments. It's like I'm a real scientist. I have also gotten pretty good at all aspects of PCR. Yesterday, I loaded a gel after PCR, and it was a real beauty! Even better, it showed that everything worked very nicely.
Hampshire's only molecular/cell biology class with lab is called Gene Cloning. It is every January. It is a three week, 8 hours a day, intense lab boot camp. My thoughts: how could they possibly learn anything useful (read: do any experiments well) in such a short period!? I have been at this now for 7 weeks, and I work eight hour days! I am only starting to get good at some things.
Today and tomorrow are the Spring Research Festival. I went and explored. It was awesome (in the quite literal sense- I was filled with awe). Think huge circus tent. Or exhibit hall if that suits you. Eight rows of tables. Easily 50 tables long. It was huge. If you register, you get a free t-shirt upon entering. It's a nice one too. I thought the easiest way to approach this was to start on the right and work my way down the tent. It seems that this is how it was organized, so it was a good choice.
Most of the tables on the right were Department of Defense, National Cancer Institute, Fort Detrick related things. This included this included childcare services, Fort Detrick's recycling program, NCI-F's library services, etc. I got a very nice pen from USAMRIID. I think that is the Army's bio-warfare science unit. It is a very nice pen though.
The next section was posters. This was mostly postdocs showing off their work. I felt bad for them. I walked down one row of posters, and it was enough. I emerged on to something that my mind had a hard time coping with.
I've only been to one kind of trade show in the past. Jewish. I have been to lots of Jewish conferences, and they often have exhibit halls. Publishers show off books, leatherers show off book-bindings, there are pretty things wrought in silver, and other pretty things woven of fabrics. A science trade show is like walking onto a movie-set for some futuristic something-or-another. There was a booth filled with really pretty tools for cutting, namely scalpels, scissors of various shapes and sizes, and tweezer-like objects. There were incubators from cute to discreet to "huh, that looks like an incubator," with sales-people showing off all their interesting features. There was a robotic pipetting booth. Basically, you use a computer program to tell it origin and destination information (and it is all color-coded and click friendly), and the robotic arm pipettes just the right amounts of the right stuff into the correct destination.
Food was brought by ZiPani, a sandwich place in town. I went there once with AK because a name like that might lead you (as it did us) to believe that it is a bread place. I got a sandwich at the ZiPani stand (it was pretty cheap and there was no sales tax!), and sat down out side at one of the picnic tables.
One of the exhibitors joined me, and his business is based in Natick MA, so he was quite familiar with Amherst, and even knew about Hampshire. He was a real pleasure to chat with. It was fun. Aside from the requisite business name and his own name, his name tag said, "Likes broccoli and is good with kids!" He was a pretty cool guy.
I came back and had to do a nuclear extraction.
A few days ago, I put some cells on cell culture plates, and transfected these cells with some DNA. This means that I inserted DNA into the cell, with the hopes that the cell would then express (manufacture) the protein that the DNA codes for.
After the cells have been expressing this protein for a couple days, I basically scrape the cells off of the plates, and put them in test tubes. The cells then get put in a centrifuge, and I separate off the liquid. I then add materials that will allow me to extract the nuclei of the cells after spinning them down again.
Today, it did not work. I noticed when scraping the cells that they were "gummy." As in, they stuck together in long strands. When I sucked them up in my pipette, they pulled like boogers. I put them in the centrifuge, and when I took them out there was no pellet. Normally when you run the centrifuge, all the heavy stuff sticks together and forms a "pellet" at the bottom of the tube. You can then suck off the liquid, and you are left with the pellet. Either you add something else at this point and re-centrifuge or you stick it in the freezer depending on what you are trying to get from the pellet.
No pellet formed. We thought that maybe I didn't spin the centrifuge fast enough, and thus there was not enough force to separate the pellet from the liquid. We spun it faster. No good. A few of the tubes looked like they might have a pellet, but when I tried to pull the liquid, the boogers came with it.
So what does this mean? It means that my cells died before I scraped them. I admit that I did not check on them under a microscope since I transfected them (two days ago). But they should have been healthy. We were using a different serum this time. Normally we put FBS (fetal bovine serum) in the media, and the cells grow very happily. However, we were out of FBS, and we used a different company's FCS (fetal calf serum). These really should be the same thing. So naming conventions aside, the difference was that the FCS was heat-treated. The going theory at the moment is that my cells were acclimated to our regular serum/media combo, and the shock of changing this was detrimental to them. Who knows? No use speculating.
So on Friday, I will start a new transfection process.
In other news, NIH has apparently censored signing in to a blogger blog. I am able to read blogs, but theoretically not post or comment. I happened to have set up a way to blog by email. Unfortunately, I can't check formatting and whatnot. Hopefully that works.
--
-Tal
Thursday, April 15, 2010
Practice makes perfect
So... my PCR from a couple of days ago kind of failed. Yesterday, I did an agarose gel electrophoresis. This is basically to see whether the PCR was successful, and it was not.
Gel electrophoresis is really cool. You start with an individually sealed package. Inside, is a plastic slab. The plastic slab houses a "gel." In this case the gel is agarose, the sugar-matrix that is made from red algae, and is used in lots of deserts and "seaweed salad" in Asian foods. Read more about it here. The gel has wells at the top. These are very small holes where the gel is cut out. You put the gel-slab rectangle into a bigger plastic box. It is covered with a buffer solution that is electro-conductive. There is a cathode and an anode (positive and negative electric terminals) on either end of the box, the DNA itself is electrically negative. Below is a picture of the slab in the electro-conductive box.

At the top, against the blue background, you can see the wells. A pipette is used to load material (in our case, DNA) into the gel.
First, it is really difficult to get the pipette right into the well, and further to put your material in there. Light is refracted against the buffer solution, and makes aim difficult. You are also using this long pole (the pipette) with a tiny end that needs to fit inside a tiny hole. Regardless, once you have the material in place, you seal the box. It is connected to wires that are connected to a power supply. It's basically a completed circuit. Electricity flows from the anode (negative) through the gel to the cathode (positive). It also pulls the DNA along with it.
Before putting the DNA in the wells, dye was added. This dye, called Ethidium Bromide, binds to each base of DNA- one molecule of dye for each base. This dye allows you to see how far the DNA ran on the gel. The farther the band, the smaller the molecule. Basically, the smaller the molecule, the faster it goes when electrocuted. The location of a band on the gel tells you what you are looking at.
When the electrophoresis is over, you take the gel to a chamber that emits UV light. This is a small light-proof box. You put the gel inside, and seal it up. You turn on the UV light, and can take a picture of the gel using a camera in the box. Ethidium Bromide glows when it is lit by UV light. The resulting photograph shows the bands where there is Ethidium Bromide and thus DNA.
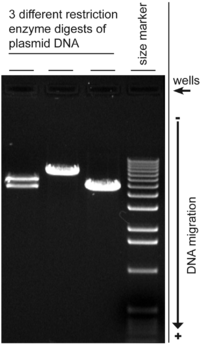
The following is a picture of the UV illumination:
The more dense a band is, the more material there is.
With the gel that I ran, shown below, the left-most column was a mistake, and is to be disregarded. The second from the left is a negative control. It was basically only dye, to have a scale. The third is the positive control. The fourth and final column was my experiment. It was looking for a region called DMR. You see that band at the bottom of the the final one? That is DMR. I found it. However, the problem is the control. It is blurred and crazy. We have no idea what exactly went wrong there.

So the DMR is good, but the control is messed up. We can keep the DMR that I made in PCR, but have to run the control again, else we won't be able to actually experiment with it.
AB decided that he would set up the control for PCR so it would be quicker, and if I noticed anything that he did differently, then we might know what went wrong.
Start to finish, he was done in 15 minutes. He did half of what I did, but when I did it, it took me an 1.5 hours. This means that he did it three times faster than me. I guess practice makes perfect.
LATER: It is now four hours after I wrote that up. AB's new PCR is done. We also ran the gel electrophoresis. We took a picture. You know what? His looks just the same as mine.
This means a number of things. First and foremost, I didn't mess everything up. The other really big thing, is that we have no idea what is wrong. We have pretty much found that it wasn't really the mixing that messed things up. It might be either the quality or quantity of the reagents that went into it. Also, it might be something with the temperature of the thermocycler.
Next week, we will try to address this problem. Possibly, there was too much template DNA. This is odd because this PCR has been successful twice before, but has now failed twice. Possibly, it means that the amount of template DNA is right on the threshold- in the right conditions it might be enough but under the wrong conditions it's too much. We have no idea what the "right conditions" actually are.
Gel electrophoresis is really cool. You start with an individually sealed package. Inside, is a plastic slab. The plastic slab houses a "gel." In this case the gel is agarose, the sugar-matrix that is made from red algae, and is used in lots of deserts and "seaweed salad" in Asian foods. Read more about it here. The gel has wells at the top. These are very small holes where the gel is cut out. You put the gel-slab rectangle into a bigger plastic box. It is covered with a buffer solution that is electro-conductive. There is a cathode and an anode (positive and negative electric terminals) on either end of the box, the DNA itself is electrically negative. Below is a picture of the slab in the electro-conductive box.

At the top, against the blue background, you can see the wells. A pipette is used to load material (in our case, DNA) into the gel.
First, it is really difficult to get the pipette right into the well, and further to put your material in there. Light is refracted against the buffer solution, and makes aim difficult. You are also using this long pole (the pipette) with a tiny end that needs to fit inside a tiny hole. Regardless, once you have the material in place, you seal the box. It is connected to wires that are connected to a power supply. It's basically a completed circuit. Electricity flows from the anode (negative) through the gel to the cathode (positive). It also pulls the DNA along with it.
Before putting the DNA in the wells, dye was added. This dye, called Ethidium Bromide, binds to each base of DNA- one molecule of dye for each base. This dye allows you to see how far the DNA ran on the gel. The farther the band, the smaller the molecule. Basically, the smaller the molecule, the faster it goes when electrocuted. The location of a band on the gel tells you what you are looking at.
When the electrophoresis is over, you take the gel to a chamber that emits UV light. This is a small light-proof box. You put the gel inside, and seal it up. You turn on the UV light, and can take a picture of the gel using a camera in the box. Ethidium Bromide glows when it is lit by UV light. The resulting photograph shows the bands where there is Ethidium Bromide and thus DNA.
The following is a picture of the UV illumination:
The more dense a band is, the more material there is.
With the gel that I ran, shown below, the left-most column was a mistake, and is to be disregarded. The second from the left is a negative control. It was basically only dye, to have a scale. The third is the positive control. The fourth and final column was my experiment. It was looking for a region called DMR. You see that band at the bottom of the the final one? That is DMR. I found it. However, the problem is the control. It is blurred and crazy. We have no idea what exactly went wrong there.

So the DMR is good, but the control is messed up. We can keep the DMR that I made in PCR, but have to run the control again, else we won't be able to actually experiment with it.
AB decided that he would set up the control for PCR so it would be quicker, and if I noticed anything that he did differently, then we might know what went wrong.
Start to finish, he was done in 15 minutes. He did half of what I did, but when I did it, it took me an 1.5 hours. This means that he did it three times faster than me. I guess practice makes perfect.
LATER: It is now four hours after I wrote that up. AB's new PCR is done. We also ran the gel electrophoresis. We took a picture. You know what? His looks just the same as mine.
This means a number of things. First and foremost, I didn't mess everything up. The other really big thing, is that we have no idea what is wrong. We have pretty much found that it wasn't really the mixing that messed things up. It might be either the quality or quantity of the reagents that went into it. Also, it might be something with the temperature of the thermocycler.
Next week, we will try to address this problem. Possibly, there was too much template DNA. This is odd because this PCR has been successful twice before, but has now failed twice. Possibly, it means that the amount of template DNA is right on the threshold- in the right conditions it might be enough but under the wrong conditions it's too much. We have no idea what the "right conditions" actually are.
Tuesday, April 13, 2010
PCR
Finally, I can sit down to have my heart-attack. For the last 1.5 hours or so, I have been setting up PCR, or the polymerase chain reaction. This is the first step in a much larger process and the first big thing that I have done by myself.
PCR is a very complicated process. The premise is to be able to copy (many, many times) a piece of DNA. There is a particular segment of DNA that you want to study, or in our case you want to study the proteins that bind to that DNA.
A bunch of reagents get mixed together. These include primers, dNTPs, polymerase, buffer, and of course the DNA template. There are forward and reverse primers, each one travelling in one direction along the DNA strand. They are complementary to the ends of the template. dNTPs (deoxynucleoside triphosphates) are the building blocks of DNA. Polymerase is the stuff that actually does the copying/making new DNA.
After mixing these together in the right proportions, which is the only thing I was actually doing for the past hour and a half, they are put into tiny thin-walled test tubes called PCR tubes. This is so that their temperatures can be changed very quickly. There are 50 micro-liters in each tube (about 1 drop of water's worth). These get put into a thermocycler. This is a device that is able to change temperature very quickly, and sustain a certain temperature for a given amount of time. Each temperature allows for different parts of PCR to occur.
First, the mixture is heated a lot. In our case, 94 degrees C. This is to denature the DNA, ie. to make it melty. It separates the bonds of the DNA that would normally make it a double spiral. It is now two single strands.
The temperature now goes down to 55 degrees C. This is the optimal temperature for the primers to attach to the strands. Remember how there is a forward and a reverse? The primer attaches in the middle of the strand, so this is necessary. Also, it can only do reverse in short segments, and these have to get zipped together at the end. Thus, the forward primers need to circle back. The polymerase will now attach to the primer-template hybrid. This is called annealing.
The temperature is raised to 72 degrees C. This is the optimal temperature for the polymerase to make new DNA. The primer is sort of a blueprint for the polymerase to know how/which dNTP to attach to make DNA. This is the extension step.
This process is one cycle. With each cycle you have double the amount of DNA material. We repeat this 45 times. I think that means we end up with 2^45 times the amount of DNA than we originally had as the template. Regardless of the math, it's a tremendous duplication.
At the end, it gets chilled to 4 degrees C, and it can stay there indefinitely. My PCR wont be done before the workday is over, so I can fetch it tomorrow.
I did this all by myself. *pats self on back, and says, "I am terrific."* AB wasn't even looking over my shoulder. Tomorrow, we get to see whether it worked.
PCR is a very complicated process. The premise is to be able to copy (many, many times) a piece of DNA. There is a particular segment of DNA that you want to study, or in our case you want to study the proteins that bind to that DNA.
A bunch of reagents get mixed together. These include primers, dNTPs, polymerase, buffer, and of course the DNA template. There are forward and reverse primers, each one travelling in one direction along the DNA strand. They are complementary to the ends of the template. dNTPs (deoxynucleoside triphosphates) are the building blocks of DNA. Polymerase is the stuff that actually does the copying/making new DNA.
After mixing these together in the right proportions, which is the only thing I was actually doing for the past hour and a half, they are put into tiny thin-walled test tubes called PCR tubes. This is so that their temperatures can be changed very quickly. There are 50 micro-liters in each tube (about 1 drop of water's worth). These get put into a thermocycler. This is a device that is able to change temperature very quickly, and sustain a certain temperature for a given amount of time. Each temperature allows for different parts of PCR to occur.
First, the mixture is heated a lot. In our case, 94 degrees C. This is to denature the DNA, ie. to make it melty. It separates the bonds of the DNA that would normally make it a double spiral. It is now two single strands.
The temperature now goes down to 55 degrees C. This is the optimal temperature for the primers to attach to the strands. Remember how there is a forward and a reverse? The primer attaches in the middle of the strand, so this is necessary. Also, it can only do reverse in short segments, and these have to get zipped together at the end. Thus, the forward primers need to circle back. The polymerase will now attach to the primer-template hybrid. This is called annealing.
The temperature is raised to 72 degrees C. This is the optimal temperature for the polymerase to make new DNA. The primer is sort of a blueprint for the polymerase to know how/which dNTP to attach to make DNA. This is the extension step.
This process is one cycle. With each cycle you have double the amount of DNA material. We repeat this 45 times. I think that means we end up with 2^45 times the amount of DNA than we originally had as the template. Regardless of the math, it's a tremendous duplication.
At the end, it gets chilled to 4 degrees C, and it can stay there indefinitely. My PCR wont be done before the workday is over, so I can fetch it tomorrow.
I did this all by myself. *pats self on back, and says, "I am terrific."* AB wasn't even looking over my shoulder. Tomorrow, we get to see whether it worked.
Friday, April 9, 2010
Really? More Bureaucracy?
First and foremost, I don't think I will ever learn to spell bureaucracy. So, thank yous are in order to the inventor, proprietors, and purveyors of spell-checking technologies.
President Bush signed an executive order that said that all federal employees should use the same wireless access card, where the only difference is the logo of the department you work for. We are currently moving to that system. In order to get this new access card, all federal employees need a background check. Two days ago, I got my email saying that I needed to complete my application for the background check. I went to a special website, and put in my identifying information, and it presented me with a huge mess of forms to fill out.
I mentioned this to AB, who said that maybe I should contact the agency and tell them that I was only interning for a few months, and maybe its a waste of resources? I called them, and they told me that I should go ahead and fill it out. That they will initiate the background check and at whatever point it was left at when I leave they will leave it. I thought that this is a tremendous waste of resources, but there is plenty of waiting time during the day, so it will give me something to do.
It was really in depth. They wanted me to account for every place that I have lived in my life, and who (name, address, phone number) could account for my being there. Relatives don't count. Further, account for every job or period of unemployment since turning 16. Again, someone has to be able to account for my being there. What schools have I gone to? Finally, give three character references (again, no family), with adresses, phone numbers, and length of time that I've known them. They should be able to account for the last five years. It was really intense.
Now, they are going to waste your tax dollars in researching that information.
President Bush signed an executive order that said that all federal employees should use the same wireless access card, where the only difference is the logo of the department you work for. We are currently moving to that system. In order to get this new access card, all federal employees need a background check. Two days ago, I got my email saying that I needed to complete my application for the background check. I went to a special website, and put in my identifying information, and it presented me with a huge mess of forms to fill out.
I mentioned this to AB, who said that maybe I should contact the agency and tell them that I was only interning for a few months, and maybe its a waste of resources? I called them, and they told me that I should go ahead and fill it out. That they will initiate the background check and at whatever point it was left at when I leave they will leave it. I thought that this is a tremendous waste of resources, but there is plenty of waiting time during the day, so it will give me something to do.
It was really in depth. They wanted me to account for every place that I have lived in my life, and who (name, address, phone number) could account for my being there. Relatives don't count. Further, account for every job or period of unemployment since turning 16. Again, someone has to be able to account for my being there. What schools have I gone to? Finally, give three character references (again, no family), with adresses, phone numbers, and length of time that I've known them. They should be able to account for the last five years. It was really intense.
Now, they are going to waste your tax dollars in researching that information.
Thursday, April 8, 2010
journal access
One of my biggest upsets/pet-peeves at Hampshire is the library's access to journals. You do some pretty great searching, find the articles that you NEED, and cross your fingers. Praying to whom/whatever does not seem to help. Occasionally, that article that you need is accessible at Hampshire. More often than not, it's not. Hampshire doesn't have all the money that many more established colleges might. As such, we don't have the library services that other colleges might. Often this can be remedied with a trip to UMass down the road or through inter-library loan (you can request a journal article that we don't have, and usually within a week, a photocopy of it magically appears for your higlighting pleasure). What if it just worked though? What if you could do research, and simply click, and there it is?
I admit I didn't know what I was missing. I was able to deal with the planning ahead that is required in doing research at Hampshire.
The NIH has subscriptions to everything. Just click, and there it is. In PDF, HTML, or other formats. From multiple sources. It's just so easy. And crazy expensive. Again, your tax dollars at work.
I admit I didn't know what I was missing. I was able to deal with the planning ahead that is required in doing research at Hampshire.
The NIH has subscriptions to everything. Just click, and there it is. In PDF, HTML, or other formats. From multiple sources. It's just so easy. And crazy expensive. Again, your tax dollars at work.
Monday, April 5, 2010
The point (sorry, but Ringo is not here)...
I have finally found out the point of what we are doing. It's about time. Until now, I have been doing a whole series of processes without really knowing how they relate to each other, or their end purpose.
We are both a "Core Services" lab and a "Research and Development" lab. Core Services mean that scientists around the country can hire us to do experiments for them. The R&D bit means that we aim to design new technologies and techniques for understanding proteomics. Most labs are one or the other. Both together though works very well. This means that scientists can work with us to design an experiment that will create a new technique or give new understanding in an area.
Apparently, the project that AB and I are working on has immediate applications in cancer chemotherapy. One of the problems of chemotherapy is the toxicity of it. One of the reasons for this is that chemotherapy affects how cells reproduce (at a DNA level). Many chemotherapies, in addition to targeting cancerous cells/reproductions, also inadvertently target the DNA repair mechanism in healthy cells. In a normal healthy cell there is an RNA protein machine that repairs nicks in the DNA. It sort of slides along the chromatin and unzips it in places that need to be repaired. The chemotherapy, however, causes this unzipping to get stuck. The RNA protein machine then sort of tacks on the end of the DNA. If this happens only a couple of times, generally the cell can survive (and this does happen occasionally in healthy cells to no detriment). Unfortunately, if it is continued, it is toxic to the cell. Thus one of the reasons of chemotherapy toxicity. We are looking at interactions of a particular protein with this RNA protein machine. It seems that in vivo (in the actual cell), this protein that we are studying will actually cleave the RNA protein machine off the end of the DNA allowing the DNA to return to normal functioning. If this turns out to always be the case, this protein could be infused with the normal chemotherapy for a cocktail that will reverse the damage that chemotherapy causes.
Here's the thing. I don't believe that it is that simple. It is amazingly difficult to study protein interactions that involve more than say three proteins (and even that many is pretty difficult). While it may be true that the protein that we are studying may be part of this interaction, there is no telling yet whether it is directly responsible for those results. Even so, it's some pretty cool revolutionary stuff.
We are both a "Core Services" lab and a "Research and Development" lab. Core Services mean that scientists around the country can hire us to do experiments for them. The R&D bit means that we aim to design new technologies and techniques for understanding proteomics. Most labs are one or the other. Both together though works very well. This means that scientists can work with us to design an experiment that will create a new technique or give new understanding in an area.
Apparently, the project that AB and I are working on has immediate applications in cancer chemotherapy. One of the problems of chemotherapy is the toxicity of it. One of the reasons for this is that chemotherapy affects how cells reproduce (at a DNA level). Many chemotherapies, in addition to targeting cancerous cells/reproductions, also inadvertently target the DNA repair mechanism in healthy cells. In a normal healthy cell there is an RNA protein machine that repairs nicks in the DNA. It sort of slides along the chromatin and unzips it in places that need to be repaired. The chemotherapy, however, causes this unzipping to get stuck. The RNA protein machine then sort of tacks on the end of the DNA. If this happens only a couple of times, generally the cell can survive (and this does happen occasionally in healthy cells to no detriment). Unfortunately, if it is continued, it is toxic to the cell. Thus one of the reasons of chemotherapy toxicity. We are looking at interactions of a particular protein with this RNA protein machine. It seems that in vivo (in the actual cell), this protein that we are studying will actually cleave the RNA protein machine off the end of the DNA allowing the DNA to return to normal functioning. If this turns out to always be the case, this protein could be infused with the normal chemotherapy for a cocktail that will reverse the damage that chemotherapy causes.
Here's the thing. I don't believe that it is that simple. It is amazingly difficult to study protein interactions that involve more than say three proteins (and even that many is pretty difficult). While it may be true that the protein that we are studying may be part of this interaction, there is no telling yet whether it is directly responsible for those results. Even so, it's some pretty cool revolutionary stuff.
Monday, March 29, 2010
Value
We use a lot of stuff here. Everything is "disposable," and should be disposed of. Alright, not everything, but it sure feels that way. There is a lot of glass that is one time use. Glass is expensive right?
Two types of pipettes (only two that I am aware of!) are used for measuring small volumes. A pipette is basically a turkey baster but much more accurate and precise. The first type has a dial that you use to tell the device how much it should suction. This type works mostly in microliters. Do you have any idea how small a microliter is? There are about 40-50 microliters to a drop of water (depending on pressure, temperature, etc.). So we pull up 10 (or some such number) microliters in this pipette and put it in a different container, like a tube. Every time that we are introducing a new reagent (ree-agent. Fancy word for ingredient), we need a new tip for the pipette. You draw up a bit in that tip, eject the tip in the disposal container, and grab a new tip. We go through at least 10 of these tips a day, and as much as 50.
The second type of pipette is for larger volumes. For amounts of liquid that are larger than 1 milliliter (ml), we use this second type. There is a handle that is sort of like a hand-gun. It has two triggers. One pulls liquid up, and the other pushes liquid down. This is done by way of a pump mechanism in the handle. This pipette also has tips. However, each tip is a long glass tube of at least 10 inches in length. These are marked along the side with ticks for varying amounts of volume. You could put in a 50 ml tip or a 10 ml tip or anything between. These are not nearly as accurate as the smaller volume pipettes because you are using your eyes rather than calibrated machinery. These are used when it is OK to be approximate. Whenever a "recipe" calls for 10 ml of this reagent, it means around 10 ml. These tips that are specially calibrated and marked (and have a filter at the top to make it harder to destroy the handle by pulling up too much liquid) get disposed of after each use as well. I imagine they are the more expensive type as they are glass.
We also use a type of non-calibrated, non-filtered glass tip. This is to get rid of waste liquid. You attach it to a vacuum tube and it sucks away liquid stuff. Each of these has to be disposed of after use or if you change the type of liquid you are vacuuming.
These latter types get used whenever you are doing tissue culture, as you are working with "large" amounts of liquid, and have plenty of waste.
There is also the issue of gloves. Every time you enter a lab it's a new pair of gloves. When you leave (even if you are coming back), you throw them away.
Then the cost of reagents. A lot of things are for general use and come in large quantities. Sometimes you get something that is at a high concentration and you only use it at low concentrations. It lasts a while. Sometimes however, you need something that is very particular or of particularly high quality. A little vial that has 1 ml (20 drops of water worth) in it can sometimes cost as much as $300 (probably even more than that). Even the more common, bulk ones are not so cheap. A half-liter of PBS (Phosphate Buffered Saline), a reagent that we use quite often, is more than $30. We generally use 10 ml per plate. If we are doing 20 plates, that is 2/5 of a bottle. Trypsin, which is used in pretty much everything, is $40-$100 for 100 micrograms, depending on the grade (the lower grade can be used for many things).
Gels, which are used to see whether the proteins or DNA you were looking for are actually present, run upwards of $40 each. Ones that are actually useful for what we do run upwards of $60 each.
I have not gone into any of the one time costs such as the initial investment in a piece of machinery. Nor have I gone into utility cost (I'll let you guess; hint- it's astronomical).
What I am trying to get at here is that science is expensive. I probably think about the cost too often. I am afraid of how much I cost, in that I am not experienced and therefore my work is not as efficient or correct as a more-well-trained individual. I will keep on trying and spending your tax-dollars though!
Friday, March 26, 2010
Goals
The lab I am with is the Laboratory of Proteomics and Analytical Technologies. Particularly the proteomics part. If the genome is the sum of all DNA and gene interactions, the proteome is the sum of all protein interactions.
DNA is the code that tells the cell (and further, the body or organism) how to make proteins. Proteins are vastly complicated. There are only 20 amino acids, which act as the base units for making protein. A protein is just a string of amino acids folded into a particular way. Proteins can be strung together in interesting ways to make protein machines. All of the action, anything that is dynamic, in a biological system can be traced to proteins. For example, digestive enzymes are type of protein that breaks apart food stuff so that it can be digested better. Lipase is a digestive enzyme that breaks fats down into triglycerides, which is how the body utilizes fats. Lipase is a protein with a special function. By educated estimates, there are thought to be over 50,000 proteins involved in making humans work properly (and improperly).
There are proteins that stay in the cell nucleus (where DNA is located). Some of these help with DNA copying, or in making other proteins. Many of them bind to DNA at certain locations. That is what we are currently studying.
There are about 10,000 employees of the NIH on this base, so my group (less than 10 people) is studying a very particular piece of the pie.
Proteins are very small. You can't see/identify particular proteins in a microscope. The best method we have currently for looking at proteins is very indirect. It is called mass spectrometry. For this method to work, you need to have a sample of proteins. These are given an electrical charge. They are also cut up. These charged pieces are sent through a constant electrical current from one electrode to another that has a sensor. We are then able to tell the size and mass of these pieces by seeing how long it took them to get through the current. By putting them all together, we are then able to see which proteins were in the sample.
While we are a mass spectrometry lab, we sort of out-source the mass spec to the experts. We design and do the experiments that get the proteins ready to be looked at in the mass spec.
So what are we looking at? We are particularly interested in chromatin and the proteins that bind to it. Chromatin is the fibers that DNA is supported on. When I say DNA, you think double-helix, right? The DNA itself is the rungs on that ladder. The actual structure, helical part is chromatin. So, the long term goal is to develop a way to look at a specific part of chromatin, and the proteins that bind to it. Current research generally looks at the protein complex without DNA, but that is likely an important part of the picture. It seems that some proteins identify where to bind because of the DNA sequence at that location, and that part of its ability to bind is based on DNA as well. This can be illustrated by thinking of a house you want to visit. You find the particular house by its address, but you don't know where to enter until you find the door. By looking at chromatin and the proteins without the DNA, it is as though you visit a house (you've followed very detailed directions to get there) and are inside, but someone studying you from above makes the house invisible to see you standing there in what was the kitchen. If you are holding a frying pan, it might be clear as to where you are. If you are simply sitting down, you might be at the kitchen table, in the living room, or even on the toilet.
Looking at protein-binding on chromatin out of context is like this. We don't know whether that is the location that the protein would normally be if the DNA was still intact or whether the interactions are more or less as complex in vivo.
The intermediate goal is to be able to understand some or all protein interactions through a model system. Also, to learn novel things about proteins that we already think we know about. One issue is what is called "background." If we are studying p53 (a human protein that we are actually studying), there are bound to be tons of other stuff that just happened to be chilling around there at the time that we harvested the sample. How much of it is supposed to be there and how much is non-specific? In the past, background has been washed away, but then you also lose some of your sample. And it's hard to know how this affects the way you would find interactions in vivo. I don't know how we are trying to get around that.
Those are some of the goals of the group I am working with. It's hard to know how to actually get there, but basically you change little things until it gets you closer.
If you want to learn a bit more, the wikipedia post on protemics is pretty helpful as is the proteopedia.
science
I realize that up until this point, I have not really mentioned much about science or even that I have started working in the lab. I started working in the lab on Monday. This is the end of my first full week. It has been such a learning experience.
I have not really mentioned anything about the science for fear of not being able to make myself understood. Here is my charge to you: if you don't understand, please let me know! I will not get better at making science accessible unless you tell me where I have assumed things that are not true.
Also, bear with me. I don't totally understand it either. I would ask questions, but I am not sure what questions will make things clearer. My next post will be all sciency. Please help me to explain it better by asking about things you don't understand. Thanks!
Thursday, March 25, 2010
JK comes out of his shell
Apparently, JK is quite a character. He asked me last night whether I had plans for the evening, I didn't so he invited me out to a local brewery with a friend.
The brewery has a deal on weeknights that from 8-12 beers are half off. They also put out a free wings buffet. SH, whom I have met on a couple of occasions, was there when we arrived. He had managed to get us a table (I have never seen a bar so busy on a weeknight!)
It turns out that both of them are politically liberal, which I had not known until this point. We discussed the healthcare overhaul mostly. SH does not stay too on top of the news. He did not know most of the details of the new law. For instance, he thought that health care would now be free. JK and I made it all clear to him.
Another big topic was eating contests. JK told us about a burger place in PA where you can get a 1, 2, 3, 10, or 30 pound burger. The two and three pounders were contests. You have an hour to eat it and if you do, the meal is comped, you get a gift certificate, and a little plaque on the wall.
A couple of years ago, JK gained about 10 pounds. He started this off by saying that he gained a bit of weight, to which SH responded incredulously, "A little?!" JK's response to the situation was, "Fuck it, I'm gonna eat what I want." It was a few month's until New Years, and JK wanted to gain as much weight as possible before the New Year. He was about 185 (whereas previously he had been about 175). He wanted to pass the 200 mark before the New Year. He would eat until it hurt. All the time. On New Years eve, he finished it off with 5 junior bacon cheeseburgers and a four pack of chicken nuggets from Burger King. His weight was 203. He then went on a diet and started working out. He is now in pretty good shape and goes to the gym (with SH or his girlfriend or both) two to three times a week.
It seems that a bar is JK's natural habitat. Previously I had thought he was kind of insecure and without much character. He totally opened up, and it was great.
Wednesday, March 24, 2010
Parking v2
Apparently the decal just works. I drove on to the base this morning, handed over my ID, and he handed it back and told me that I should have a good day. Much easier and quicker.
Tuesday, March 23, 2010
Parking
I lied a little when I said that the bureaucracy was finished. Only this morning did I get a parking decal for my car. Previously, I could only enter at one gate of the base, come at least 15 minutes early, and get in the non-decal line of the security checkpoint. When it was finally my turn, they would take my license, and write down a lot of information. They would ask me to open up everything in my car. This includes center console, glovebox, all doors, trunk, and apparently the fold-down arm rest in the back. Then, they go around the car with this funny mirror that is basically a much larger version of a dentist's mirror. It allows them to see the entire under-carriage of the car. They reach under the seats, and feel around. Then they tell me that I can close everything up, present me with a dashboard pass, and I can then find parking. Now I have a decal. I can enter any of three gates, I drive right up, they ask to see my NIH ID, and that's it. Theoretically speaking. I'll let you know if that works tomorrow.
Monday, March 22, 2010
A dip of the toes
When I went in on Tuesday, I was not expecting to stay there for a whole work day. In fact, I thought I would do my blood test and get my ID, then go home. I got to stay the whole day and it was great.
TA and AB collaborated in creating a list of papers for me to read. They gave me 12. I don't know if you have ever read a scientific paper, but there are really two types: easy to read/understand or is-this-English-it-is-not-comprehensible-at-all. They were all the latter type. Unfortunately, very few of the classes that I have taken at Hampshire were of any use to me in making heads or tails. I thought that maybe if I just read them, it would eventually click. Sort of like immersion in another language. So I just kept chugging through. I would finish a paper and think, 'I have no idea what their goal was, or if they accomplished it!'
For the past few months (since the beginning of January), I have been teaching myself Cell Biology out of a textbook. CJ told me which chapters I should definitely cover, and which I could skip. Unfortunately, he told me I could skip a lot of the protein and DNA stuff, which is all that these papers were about. I did preempt this though. I was interested in the protein and DNA stuff, so I did not skip that much of it. CJ, I know you meant well, and I don't hold it against you.
I wrote down questions I had about the papers. I was not sure that any of them would be particularly helpful, but I asked things about what a certain technique did, or why was it important to do such-and-such? I spent the entirety of Wednesday reading papers (I had spent some time reading papers on Tuesday as well, in between bureaucratic nonsenses). TA said that tomorrow (Thursday), I would meet with AB and him to ask questions and have a learning session. He set aside 1.5 hours, and I got nervous. We would meet at 11.
I came in on Thursday, and sat down to go over papers. And it clicked. I was not reading jargon, but I mostly understood what was going on! I finished that paper in probably half the time it had taken me to do earlier ones. [If anyone ever tells you that the human brain is not good at adapting, you should laugh at them...]
I finished the other papers I had left pretty easily, and went in for my meeting. I brought a sheet of questions, and even took notes. I did not need to ask half of the questions at that point. I think that TA and AB were impressed with my grasp of the material (they had not intended that I finish all the papers before Friday). They told me what they intended for next week, but they were in between experiments, so there was nothing for me to do in the lab until Monday. I asked if there was any reason for me to come in on Friday, and they could not see any (I am paid salary, not hourly, so if they said I could be off, I was still paid for it). It was not even lunch yet, so I went home and made lunch.
AK and I then went out to a park, and went on the swings a bit. I got to hang out with her for the rest of the weekend too! It was great. We went to a mega-church on Sunday morning. AK is studying fundamentalist Christianity, so it was a real treat for her. She really enjoyed it. I enjoyed it too. It is a really well-oiled machine! They gave us some really great travel mugs because it was our first time there.
AK had to leave shortly after the service so that she could be back at Camp Hamp before the vampires came out. I miss her already.
Bureaucracy!
Bureaucracy. The best word to describe the NIH and the NCI in just one word is bureaucracy.
I'll rewind a moment here. I admit it has been a while since I wrote anything. Sometimes it felt like it was worth writing, and sometimes it felt like there was very little new information.
I went into the office on Friday, March 5, and did paperwork. An hour of paperwork. In fact, there was a checklist of the paperwork that I had to complete before EOD (Entry On Duty). Poor DM, I sat at his computer and filled out my paperwork. Apparently, all the forms are online, but your computer has to be approved before you can access them. So I filled out a personal data sheet and a medical history sheet and bank direct deposit sheet and various others. I was then introduced to LC, the secretary for this part of administrivia at SAIC. She really is a sweetie. We had a sort of implicit agreement about the ridiculousness of all the paperwork. LC told me that my medical history needed to be signed by my primary care physician. My direct deposit form had to be signed by the bank. The physician was easier- I sent them a fax and asked that they sign it. It took a few days, but it came in. For the bank, I called the bank (which only has a physical presence in Texas) and told them about the direct deposit thing. They immediately faxed their own direct deposit form with a signature on it. Not good enough for the US government. It has to be their form, even though both had all the same information! I called the bank back, and they said that it might take three business days to deal with this particular form. Grr. When I had done all I could there, DM told me about an event that evening. They were having a going-away party for one of his post-doc fellows, and I was invited. He told me how to get to the restaurant, and even printed out something with the address and a review.
I went back home. JK had a lot of trouble understanding that I had gone into work for less than two hours and was already back... He works from home and is plainly aware of my comings and goings. I hung out and did very little. I looked at my clock and realized that I was already a half hour late for the dinner thing. DM had said that he was planning to be at least a half hour late, so I figured I was still pretty safe. I went. I only got a little lost getting over there, but my phone corrected me (if you have not heard me praise the Android phone operating system from Google, this is the first such praise). I got to the restaurant, which appears to be a converted mansion. It was quite large. I did not see DM, but I saw LC. She apparently works there as a second job in the evenings as a hostess. She pointed me in the right direction. DM was happy to see me. He introduced me to everyone around the table including his 10-year-old daughter. Whenever he felt appropriate, he would turn to me and explain something someone had said, or about some relationship. He wanted to make sure that I understood the inside jokes. It was pretty welcoming. Eventually, he had them bring in the bill. He wouldn't let anyone help pay. I was kind of glad of that myself, but the colleagues were miffed. Apparently, he has pulled this stunt before, and no one likes it. Hell, if my boss wants to pay for my dinner, who am I to complain? He left with his antsy daughter, and I learned that all the staff love him. They invited me to karaoke on Tuesday night. I neglected to ask details, but DM had explained earlier that the karaoke bar is this super redneck affair. You walk in there and realize that you have more teeth than three other patrons put together. It sounded exciting to me.
DM was going to be in Bethesda on Monday, so I should call Tuesday to come in for preliminary blood tests and getting an ID badge. I called Tuesday, and he said that LC had said that they were still waiting on things to process so I should come in on Thursday. I asked about karaoke that night, and he said that thought it would be at Cactus Flats, but he did not know a time. I figured that 8 PM was pretty much a good time for karaoke on a weeknight. I got to Cactus Flats around then, and only saw rednecks. I left, my urge to socialize not fulfilled.
Hampshire spring break started that Friday (March 11). AK decided that she was going to miss class on Wednesday and Thursday and come down to visit on Wednesday. The original plan had been that she would leave around 5 AM, and get here probably around 3 (Google Maps said it was a seven hour trip, but she figured it would take her eight). I got up fairly early. Not actually early, but for someone that had no real reasons to be up for quite some time, 9:30 felt early. I took a shower, made some breakfast, saw JK. He asked if I had anything planned for the day, and I reminded him that AK was coming to visit. He asked about when she would be showing up, and all seemed reasonable. Suddenly, I got a call from AK. I figured she would tell me that she was taking a pit-stop, and would be here in a few hours. Instead she said, "So, I think I am parked outside your condo." It was before noon. I went out, and there she was. Apparently she had trouble sleeping the night before, so she just left around 3 AM. I hung out with AK for the day and showed her around.
I came in on Thursday. Believe it or not, I filled out more paperwork. Also, the bank still had not faxed back the direct deposit form (nor had school sent in proof of enrollment, but I expect Hampshire to be a little slow). I called the bank, spoke to three different people of varying hierarchy levels, and the last one said that she would send it in just a few minutes. Is there anything else that I would like to do? She noticed that I don't have car insurance in my name on my account, and would I like to transfer the insurance to my name and my account? No thanks. I will let my parents pay for car insurance as long as they are willing to do so. The fax came in, and I was pretty well set up. I should come back in on Tuesday to get my blood drawn and picture taken.
LC sent me an email on Monday with more bureaucracy. I needed to complete an information technology security course online. It actually took about an hour, but I learned all about how to make awesome passwords and that I should not share them with anyone.
I came in on Tuesday, March 16, to get my health inspection. I would say it was a physical, except it was not. I never saw a doctor, but hung out with the same nurse for about 20 minutes. Both when I was making my presence known for the appointment and when the nurse was verifying my identity, they REALLY wanted me to have an NIH employee ID number. I did not have such a thing. I gave them permission to store some of my blood, so that if I came in contact with any blood-born pathogens they would have a comparison. They also ran a test to make sure that I had significant immune-system defense against hepatitis B; DM had apparently written that I would be working with human cell lines and would therefore be in a high(er)-risk environment. That was new to me.
LC scooped me up from the Occupational Health Services, and brought me to the ID badge room. I presented my license to prove my identity, and the clerk asked me to take a seat. I figured I would be waiting, but she said, "Tell me when you are ready to take the picture." I said that I was, and she asked me to look at the camera. There was a black box upon a tripod near the ceiling that had been indicated. I had figured it was a surveillance-type video camera. I half-smiled while trying to look natural (it is so difficult to take a good ID picture!). Moments later, she handed me an ID card! It was quite cool and quick. And the picture came out fairly well aside from a bright white light spot on my forehead.
LC took me out to a table in the lobby area that had vinyl ID holders and various lanyards. I picked a lanyard, grabbed a vinyl cover, and then she took me to the building that my lab would be in. It was an outdoor building maze getting to that building. When we got there, the door that she wanted to use was in a hard-hat under-construction area. We found a different door, and it had a sign that said, "For access to this building see Protective Services." She tried her ID to open the door, no luck. She told me to try mine. I did, and it worked. Pretty cool.
She introduced me to SW. SW does administrivia for this building. I think she might be considered a lab manager. SW handed me a piece of paper that had my initial login credentials. The two of them, LC and SW, walked me to my workstation. It had not even occurred to me that I would have a computer space all to myself. Actually the space I was in was much more office-like than [my mind's idea of] lab-like.
LC bid me farewell, but told me that there was still plenty more bureaucracy to deal with, and that she would email me. SW had me log in and get situated slightly before introducing me to TA. TA is apparently the head of my lab or something. They have not made titles well known to me. He has an office all to himself (whereas there are six other people in the room I am in), so he must be important. He fetched AB, who it turns out is pretty much my mentor. So the scientist hierarchy is something like: Tal works under the direction of AB, who directs a project under TA, who reports to DM, who know longer actually does science but directs it.
TA and AB had a little interview with me in TA's office. It was a little bit awkward. I could not see both of them at once because of the location of the chair I was directed to sit in; I had to look left to look at TA, and look right to look at AB. They were asking about how I got where I was at that moment (I did not direct them to read this blog...), what is my major, that sort of thing. They were trying to understand how much experience I (hadn't) had. I actually noticed TA's outfit half-way through. HE was wearing a very nice sweater, pretty nice non-worn blue jeans, and stylish black velcro shoes. Each one looked good in its own right, but they did not really go together. At some point, TA said something like, "Yea, we did not know you were even a possibility until DM told us this weekend." !!!! I was a little aghast. But, then I thought about DM and it made plenty sense. AB made mention that he could not understand how science was done before the internet. TA said that you would go to the library, pull a whole bunch of journals of the shelves, look up keywords in the index, read the abstract for the article, read the references, etc. I agreed with AB. It is so much faster now. TA said that when he was in grad school he had a professor who said, "You kids these days. You don't know how to do real science. You just know how to use kits!" TA responded, "But at least we get things done and learn new things. Your generation just made excuses for why experiments did not work."
We finished the interview with TA saying he would assemble a bunch of journal articles/scientific papers for me to read over the next few days so that we would all be on the same page. And that TA would show me where to go for lunch and would eat with me.
We went to lunch, and the cafeteria is actually pretty nice with reasonable prices. We both had buffalo chicken wraps, his with chips and mine with pretzels. We chatted for a bit, and I got to know him. He is from Iceland, did undergrad there. Did grad and post-doc in the US with his post-doc appointment at the NIH. He went back to Iceland, and came back here (to the NIH/NCI) less than three years ago. He seems like a great guy, and really wants me to ask questions and to learn a bunch during my tenure here.
The rest of the day was spent back in bureaucracy land. I had received the email from LC, which said something like, "Welcome to the NCI! Here is what you have to make sure you have done:" A whole welcome site full of fill this out, take this course, do this orientation. I did an ethics training course online that took about an hour. I did an orientation to the NIH that also took about an hour. I have to admit that these online courses are really well made. You have to answer questions so that they are sure that you understand the material. Finally, five o' clock came, and I left. No one mentioned that they were going to play Taps. I left my building at about 4:55, and proceeded to my car. There were military folks and contractors (I am on a base) talking jovially about the end of the day. Suddenly, a base-wide PA system blew out Taps. For those of you who do not know, Taps is tune often played at flag ceremonies, funerals, and other formal gatherings. It is very solemn, and is most often a closing. You may also know it by the name Vespers (used in Scouting) or Day is Done, which are the first words of the second verse. When the PA came on, everyone around me paused mid-word, faced in a particular direction, and stood at attention. I did the same. We waited for Taps to end (they did at least two verses), and I went home. Thus ended the bureaucratic saga.
Thursday, March 4, 2010
Flurry of activity
It's been an exciting couple of days. Three, really. I called DM from BWI on Monday, after getting off the plane. I got voicemail, and left a message that was something like, "I am leaving BWI. I could be at the lab within an hour and a half. Call me back so I know what to do."
I drove back, and thought that I would be able to do it without a map or gps or directions, etc. I was wrong. I got a little confused about I-370 and I-270 and I-70. But that's what these new high fangled phones are for right?
I got back to my apartment unharmed. And uncalled. I took a nap (had been up since 3:30 am with some sporadic plane-sleeping), took a shower, took a skype call with AK. I finished the third season of Six Feet Under.
On Tuesday, I went into DC. I visited with a friend who goes to school at American, and we had some Chinatown Chinese food for dinner. I then went to a concert without her. She had been invited, but could not make it.
It was Hadag Nachash. Quite enjoyable. This was the fourth time I have seen them live, and it was easily the best. This was their last stop on their tour, and they really made it count. The venue was really weird. It was the Sixth and I Historic Synagogue. As in, it used to be a synagogue. Now, it is a venue for Jewish cultural programming. Sort of like a JCC, but they really know how to put on a concert. The concert was co-sponsored by J Street (the mommy organization of J Street U), so I got a free ticket and reserved VIP seating. Pretty cool. It was just weird to be at this hip-hop concert in a sanctuary.
Somewhere in the meantime, DM called. He said that he had been to the office of Mr. Guy-who-makes-decisions, and said that he needs this decision (of me being able to start immediately) today! Mr. Guy wasn't in. His secretary said she would have him call when available.
Wednesday was mostly shopping. I went to Ollie's Bargain Outlet. They are so awesome. They buy up overstock and outlet stuff. They sell it cheap. I think Ollie's should always be the first stop, because you never know what they are going to have. Afterwards, you can go to K-Mart or Walmart or the grocery store. They did not have everything I wanted but were close.
I called DM on my mother's wishes and told his voicemail that I would sit in Mr. Guy's office if need be. I only have time. Also, if DM were to give me his secretary's number, I could stop bothering him directly.
In the evening, I went to Aikido for the first time in Frederick. It was like going to Aikido for the first time ever. I have been doing Aikido basically for five years over the past seven. It has not been particularly contiguous, but I am certainly not a new beginner. I have now studied three styles, and this was a fourth. Aikido is a martial art wherein there is a lot of throwing and joint locks and that sort of thing, but it is all about compassion to your opponent. You never attack, just redirect their energy.
This dojo (training space) belongs to the Ki Society. I have always wanted to try this sort of Aikido, as they focus on cultivating Ki. Ki is life-force. It is a very difficult concept for Westerners. Basically, all of your strength in this type of Aikido actually comes from relaxing. I was working with the teacher when we were all paired up, and everytime that I would begin to use strength, he would freeze up. It was aggravating and motivating. I think I will stick with it.
This morning, DM called. He said, "Why don't you just come in tomorrow morning and we'll get you started. We are still waiting on Mr. Guy, but that's just a formality. Some come in tomorrow to my office, and we will get you started." OK. Cool. Sounds good. Etc.
I have showered and read a bit since then. He just called back (while I was writing this), and said "Mr. Guy called me. Everything is approved. You are set to go. Come in tomorrow morning, and we will get started!" Stupendous! Awesome! Suddenly I feel as though a gigantic weight has been lifted from my shoulders. And it's only lunchtime! What else might the day hold?
Monday, March 1, 2010
SPA conference round-up
I came to the Student Peace Alliance conference without any idea of what to expect. It seems that I assumed it would be mostly Jewish people. This was due to all of my previous conference experience; I don't think I ever before attended a conference with no Jewish undertones. It turns out that there were Jews aplenty, it just was not a Jewish conference. I think I can pretty much sum everything up by discussing my interactions with different persons or groups. They are in no particular order.
Aaron: The executive director of SPA. A really cool guy. I guess that part of my Jewish assumption was that Aaron mentioned to me before the conference that he wanted to have a minyan for a prayer service on Saturday. I did not actually happen. He was a little too busy to try to deal with that. He was my first contact with SPA, and he convened The Meeting. He is deeply spiritual, very into his Judaism, very down to Earth, and so willing to laugh.
Julia: The managing director of SPA. A fantastic public speaker. She was able to speak for a half hour with no notes! And clearly. I was very excited to attend a session for learning to speak well that was facilitated by her. Rightly so. Not only is she a good speaker, but a good teacher of that art. Whether or not I actually learned remains to be seen. She was the chief logistical person, and things went so smoothly. On Saturday, we got behind agenda by a half hour, and somehow she recovered that time and we finished on schedule!
Zana: One of the few students with whom I actually connected. Apparently SPA thinks of students in a different fashion than J Street U. SPA is really for anyone from middle-school to post-school community organizers. Zana got a scholarship from her school to come. She has never done peace organizing or advocacy. She had a lot of prior knowledge regarding domestic violence and other issues, but this conference was her first affirmation that she is not crazy; there are other people who think and feel in the same way she does. She is a little too sarcastic for her own good, and does not yet know how to use it only in the right situations. It was kind of exciting to see her change from the beginning to end of conference.
Mary Ann: A sweet middle-aged lady from West Texas. Her son was the lead coordinator at Southwestern University (the hosting school). She is very aware of just how rural her surroundings are, yet she has remained quite liberal. She is a lawyer who used to work with artists but now represents big oil tycoons after her husband insisted she get a "real" job. She took Kobi, a few others, and me (I was the youngest) out for drinks Friday night. She took us to a restaurant that was easily four stars, everyone got at least one round of drinks, we shared a cheese plate. The bill was probably over $100, but she covered the whole thing. She was really wonderful, and always had a smile on when she saw me.
Kobi: Once upon a time, I had no experience with planning events at Hampshire. Over the past 2.5 years, I have gained a lot of experience in that area. The first event that I helped coordinate was to bring in a speaker who had served in the Israeli Army and was brought up in a racist-against-Arabs youth movement, but who had renounced all that while in service. He is now a peace activist, and goes on speaking tours all over the country. His name is Kobi. Kobi was at this conference. It was really awesome to catch up with him. I think he was really happy to see how much of a peacenik and activist I have become. He went out with us for drinks, and in true Israeli fashion ordered a cucumber salad. We then got to talk about how awful the cucumbers and tomatoes are in this country. One of my favorite topics! He had lots of interesting insight and information to share.
Ben: I was picked up at the airport in a van with many interesting traits, such as the door usually didn't close quite all the way so when we went over a bump the door-open light would come on. More interesting than the vehicle were the people in it. Ben was one such person. He works for an NGO in Uganda called Invisible Children. They do a ton to get child soldiers in the Lord's Resistance Army to defect and rejoin Ugandan society. His work is totally amazing. He was a speaker for a break-out session (as was Kobi, but they were at the same time and I have already seen/heard Kobi's spiel), and I attended it. You should immediately open a tab and check out invisiblechildren.com. WOW.
Lisa: I kept seeing this girl who looked way too familiar. I had no idea where I knew her from but she had a very Jewish look about her. Also, she was hanging out with the kids from Brandeis so it was high probability that I knew her from some Jewish past life. Turns out we went on the NFTY (North American Federation of Temple Youth) Israel summer trip at the same time, and her group often did activities with my group. It was pretty cool to connect with her.
Emma's Revolution: A fantastic music group. Their music is folky, rocky, and all-around awesome. You know those salaam, shalom, peace shirts? OK, maybe not. It's a shirt that says peace in Arabic, Hebrew, and English. It was their idea. They are great. I got a chance to chat with both of them after the conference in the exhibition hall (I was there first and foremost as a representative of J Street U). The Emma in question is Emma Goldman. They live in DC when not touring, and want to meet up for coffee at some point. Check them out at emmasrevolution.com
Ed: My first "friend" at the conference. He noticed my tzitzit, commented on them, and proceeded to tell his life story. He is from Rochester. He was there to support his daughter. She started an SPA chapter at her high school, and brought three friends to the conference. He chaperoned. Poor guy dealing with four teenage girls who were not all his own! He was really sweet. Kept referring to Reform Judaism as reformed. He showed me pics his wife sent from their Purim celebration back home. It was very sweet. We chatted multiple times throughout the conference, pretty much whenever he didn't have his hands full.
Andrew: Probably the best connection that I made. He is the executive director of the Harry Potter Alliance. They use the allegories of Harry Potter to help to create social change in the world. It's been around for less than three years, and he has over 100,000 people around the world who have volunteered under the banner of the HP alliance. He sees Harry Potter as just the beginning; he wants to mobilize many fandoms in a fight for a social change. I attended a session by him on using new social media for organizing. YouTube and related is absolutely the future of activisms. We are so behind the times! It is some good stuff. He currently has a minor obsession with Buffy. We exchanged reading lists. This looks to be the beginning of a beautiful relationship. Check out the HP alliance at thehpalliance.com
Michael: I attended a session about letters to the editor and Op-Eds. The presenter was OK. Michael was fantastic. He has ghost-written Op-Eds for various Middle-Eastern and Asian heads of state. He currently is the communications director for a congressman. He knows his stuff. He gave me his card saying that he would be happy to send me the tips he gives to his students, or to read over any Op-Ed or letter to the editor that I write so as to give suggestions.
Echo: She was another interesting thing about the van ride from the airport. She has been in grad school for a long time. It's been long enough that her beginning credits are going to start expiring next year. By grad school, I mean seminary for Jews for Jesus. She was trying to shovel the concept of a "Department of Peace" down my throat. This was my first impression of SPA folks, and I thought, 'My goodness, What have I gotten into?!' It does turn out that SPA's long-term goal is the creation of a Department of Peace at the federal level. But they are not as pushy about it as she is. Also, they wait until they know you approve before pushing the Department of Peace concept. By the time the SPA actually mentioned it, I was ready for it and could believe in it.
Delta: They lost my bag. It got left at Atlanta with my connecting flight. I filed a claim when I got to Austin, but it took them three days (!) to get it to me. It is not fully resolved yet, as they said they would give me $50 off my next Delta flight. I would rather they just refunded me the check-in fee.
Victor: I am only including him because I was amazed that I encountered three members of the International Phonetic Alphabet (Alpha, Bravo, Charlie, Delta, etc), namely Delta, Echo, and Victor. He was also in the van to the hotel. I guess he just came along for the ride or as company. He said nothing the whole drive. Near the end, Echo asked if this was my first time in Texas, and I answered in the affirmative. She then said, "Well Victor is from San Antonio." He responded, "Welcome to Texas." And that was it.
Hotel Lady: I came into the Hotel on Saturday night. The woman at the desk looked at me and said, "This blue bag behind the desk belongs to you." I looked at her dumbly. How did she know!? She responded to my slack-jawed look, "I'm psychic. No, really you are just wearing your name tag from the conference." I took my bag to my room, tried to get in. The key did not work. I came back to the desk and she said, "Your key doesn't work." It was not a question. She knew her stuff. As I was standing there, she said to another fellow, "Who's wedding was it?" He gave her a familiar dumb look. She said, "You are wearing a pin that identifies you as best man." She explained that her uncle is a police officer and it rubbed off on her.
Southwestern University: A small liberal arts college founded in 1864 (?). They have a lot more money than Hampshire. It gives me hope for what Hampshire might look like once it has money to improve facilities. They have a fireplace. Something that I have said Hampshire direly needs. Also, they have Sodexho for dining services. Like we do. Except they are a Level 4 facility (I don't know what that means); their dining commons were so much more tasty than ours, though with less selection especially as far as veg and vegan options go. Apparently hipster-ism has found its way to middle-of-nowhere Texas as well. While I actually saw a tiny number of students (they disappear for the weekend), I saw plenty of fixed gear bikes. Related to being in the middle of nowhere, when I was speaking with Aaron before the conference, he said that it was taking place near Austin. I suppose it was "near" in Texas terms. It was a full hour from the airport.
The Meeting: Aaron's vision was/is to have collaboration between a whole bunch of peace/nonviolence groups. He organized a meeting of organizations. The real alliance behind the Student Peace Alliance. This was the first such meeting of its kind. It lasted from 4:30 in the afternoon until about midnight. But it was not painfully long, boring, or slow. Really it was so good. And powerful. There was such energy in the air. I will bring it back to J Street U, and I fervently hope that we will band together with the group. Aaron aimed high, and I think we met his hopes and ran with them.
That was pretty much the conference for me in a nutshell. I got so much more out of it than expected. I still have not heard from DM to know whether I am supposed to start today....
Aaron: The executive director of SPA. A really cool guy. I guess that part of my Jewish assumption was that Aaron mentioned to me before the conference that he wanted to have a minyan for a prayer service on Saturday. I did not actually happen. He was a little too busy to try to deal with that. He was my first contact with SPA, and he convened The Meeting. He is deeply spiritual, very into his Judaism, very down to Earth, and so willing to laugh.
Julia: The managing director of SPA. A fantastic public speaker. She was able to speak for a half hour with no notes! And clearly. I was very excited to attend a session for learning to speak well that was facilitated by her. Rightly so. Not only is she a good speaker, but a good teacher of that art. Whether or not I actually learned remains to be seen. She was the chief logistical person, and things went so smoothly. On Saturday, we got behind agenda by a half hour, and somehow she recovered that time and we finished on schedule!
Zana: One of the few students with whom I actually connected. Apparently SPA thinks of students in a different fashion than J Street U. SPA is really for anyone from middle-school to post-school community organizers. Zana got a scholarship from her school to come. She has never done peace organizing or advocacy. She had a lot of prior knowledge regarding domestic violence and other issues, but this conference was her first affirmation that she is not crazy; there are other people who think and feel in the same way she does. She is a little too sarcastic for her own good, and does not yet know how to use it only in the right situations. It was kind of exciting to see her change from the beginning to end of conference.
Mary Ann: A sweet middle-aged lady from West Texas. Her son was the lead coordinator at Southwestern University (the hosting school). She is very aware of just how rural her surroundings are, yet she has remained quite liberal. She is a lawyer who used to work with artists but now represents big oil tycoons after her husband insisted she get a "real" job. She took Kobi, a few others, and me (I was the youngest) out for drinks Friday night. She took us to a restaurant that was easily four stars, everyone got at least one round of drinks, we shared a cheese plate. The bill was probably over $100, but she covered the whole thing. She was really wonderful, and always had a smile on when she saw me.
Kobi: Once upon a time, I had no experience with planning events at Hampshire. Over the past 2.5 years, I have gained a lot of experience in that area. The first event that I helped coordinate was to bring in a speaker who had served in the Israeli Army and was brought up in a racist-against-Arabs youth movement, but who had renounced all that while in service. He is now a peace activist, and goes on speaking tours all over the country. His name is Kobi. Kobi was at this conference. It was really awesome to catch up with him. I think he was really happy to see how much of a peacenik and activist I have become. He went out with us for drinks, and in true Israeli fashion ordered a cucumber salad. We then got to talk about how awful the cucumbers and tomatoes are in this country. One of my favorite topics! He had lots of interesting insight and information to share.
Ben: I was picked up at the airport in a van with many interesting traits, such as the door usually didn't close quite all the way so when we went over a bump the door-open light would come on. More interesting than the vehicle were the people in it. Ben was one such person. He works for an NGO in Uganda called Invisible Children. They do a ton to get child soldiers in the Lord's Resistance Army to defect and rejoin Ugandan society. His work is totally amazing. He was a speaker for a break-out session (as was Kobi, but they were at the same time and I have already seen/heard Kobi's spiel), and I attended it. You should immediately open a tab and check out invisiblechildren.com. WOW.
Lisa: I kept seeing this girl who looked way too familiar. I had no idea where I knew her from but she had a very Jewish look about her. Also, she was hanging out with the kids from Brandeis so it was high probability that I knew her from some Jewish past life. Turns out we went on the NFTY (North American Federation of Temple Youth) Israel summer trip at the same time, and her group often did activities with my group. It was pretty cool to connect with her.
Emma's Revolution: A fantastic music group. Their music is folky, rocky, and all-around awesome. You know those salaam, shalom, peace shirts? OK, maybe not. It's a shirt that says peace in Arabic, Hebrew, and English. It was their idea. They are great. I got a chance to chat with both of them after the conference in the exhibition hall (I was there first and foremost as a representative of J Street U). The Emma in question is Emma Goldman. They live in DC when not touring, and want to meet up for coffee at some point. Check them out at emmasrevolution.com
Ed: My first "friend" at the conference. He noticed my tzitzit, commented on them, and proceeded to tell his life story. He is from Rochester. He was there to support his daughter. She started an SPA chapter at her high school, and brought three friends to the conference. He chaperoned. Poor guy dealing with four teenage girls who were not all his own! He was really sweet. Kept referring to Reform Judaism as reformed. He showed me pics his wife sent from their Purim celebration back home. It was very sweet. We chatted multiple times throughout the conference, pretty much whenever he didn't have his hands full.
Andrew: Probably the best connection that I made. He is the executive director of the Harry Potter Alliance. They use the allegories of Harry Potter to help to create social change in the world. It's been around for less than three years, and he has over 100,000 people around the world who have volunteered under the banner of the HP alliance. He sees Harry Potter as just the beginning; he wants to mobilize many fandoms in a fight for a social change. I attended a session by him on using new social media for organizing. YouTube and related is absolutely the future of activisms. We are so behind the times! It is some good stuff. He currently has a minor obsession with Buffy. We exchanged reading lists. This looks to be the beginning of a beautiful relationship. Check out the HP alliance at thehpalliance.com
Michael: I attended a session about letters to the editor and Op-Eds. The presenter was OK. Michael was fantastic. He has ghost-written Op-Eds for various Middle-Eastern and Asian heads of state. He currently is the communications director for a congressman. He knows his stuff. He gave me his card saying that he would be happy to send me the tips he gives to his students, or to read over any Op-Ed or letter to the editor that I write so as to give suggestions.
Echo: She was another interesting thing about the van ride from the airport. She has been in grad school for a long time. It's been long enough that her beginning credits are going to start expiring next year. By grad school, I mean seminary for Jews for Jesus. She was trying to shovel the concept of a "Department of Peace" down my throat. This was my first impression of SPA folks, and I thought, 'My goodness, What have I gotten into?!' It does turn out that SPA's long-term goal is the creation of a Department of Peace at the federal level. But they are not as pushy about it as she is. Also, they wait until they know you approve before pushing the Department of Peace concept. By the time the SPA actually mentioned it, I was ready for it and could believe in it.
Delta: They lost my bag. It got left at Atlanta with my connecting flight. I filed a claim when I got to Austin, but it took them three days (!) to get it to me. It is not fully resolved yet, as they said they would give me $50 off my next Delta flight. I would rather they just refunded me the check-in fee.
Victor: I am only including him because I was amazed that I encountered three members of the International Phonetic Alphabet (Alpha, Bravo, Charlie, Delta, etc), namely Delta, Echo, and Victor. He was also in the van to the hotel. I guess he just came along for the ride or as company. He said nothing the whole drive. Near the end, Echo asked if this was my first time in Texas, and I answered in the affirmative. She then said, "Well Victor is from San Antonio." He responded, "Welcome to Texas." And that was it.
Hotel Lady: I came into the Hotel on Saturday night. The woman at the desk looked at me and said, "This blue bag behind the desk belongs to you." I looked at her dumbly. How did she know!? She responded to my slack-jawed look, "I'm psychic. No, really you are just wearing your name tag from the conference." I took my bag to my room, tried to get in. The key did not work. I came back to the desk and she said, "Your key doesn't work." It was not a question. She knew her stuff. As I was standing there, she said to another fellow, "Who's wedding was it?" He gave her a familiar dumb look. She said, "You are wearing a pin that identifies you as best man." She explained that her uncle is a police officer and it rubbed off on her.
Southwestern University: A small liberal arts college founded in 1864 (?). They have a lot more money than Hampshire. It gives me hope for what Hampshire might look like once it has money to improve facilities. They have a fireplace. Something that I have said Hampshire direly needs. Also, they have Sodexho for dining services. Like we do. Except they are a Level 4 facility (I don't know what that means); their dining commons were so much more tasty than ours, though with less selection especially as far as veg and vegan options go. Apparently hipster-ism has found its way to middle-of-nowhere Texas as well. While I actually saw a tiny number of students (they disappear for the weekend), I saw plenty of fixed gear bikes. Related to being in the middle of nowhere, when I was speaking with Aaron before the conference, he said that it was taking place near Austin. I suppose it was "near" in Texas terms. It was a full hour from the airport.
The Meeting: Aaron's vision was/is to have collaboration between a whole bunch of peace/nonviolence groups. He organized a meeting of organizations. The real alliance behind the Student Peace Alliance. This was the first such meeting of its kind. It lasted from 4:30 in the afternoon until about midnight. But it was not painfully long, boring, or slow. Really it was so good. And powerful. There was such energy in the air. I will bring it back to J Street U, and I fervently hope that we will band together with the group. Aaron aimed high, and I think we met his hopes and ran with them.
That was pretty much the conference for me in a nutshell. I got so much more out of it than expected. I still have not heard from DM to know whether I am supposed to start today....
Saturday, February 27, 2010
To Austin?
As you may know, I am on the board of a national organization. Yes, I do like starting like that so as to inflate myself. I am on the board of J Street U, a campus-based organization that seeks to bring about middle-east peace. We believe in a two-state solution to the Israel-Palestine issue, wherein the future state of Palestine has full sovereignty and coexists peacefully with Israel. We often do campaigns on all of our different campuses to try to achieve these ends. We are a daughter organization of J Street, which is based in Washington DC. They seek a resolution to the conflict from a diplomatic approach; despite the arguments of hegemony, the US is in a unique position to create foreign policy that will create real change and possibly peace in the middle-east.
The executive director found out about an opportunity for a board member to go to a conference in Texas. This conference is sponsored by the Student Peace Alliance, which so far as I can tell is seeking to get all of the campus peace groups together for ally-stuff. OK, it sounds a lot better than I let on, but I really know nothing about it.
I am on a plane to Austin right now. I had nothing better to do! I have an indefinite start date for my internship. I called DM and told him that unless he had more info, I was going to this conference. He was fine with it. They (Student Peace Alliance) is paying for everything. Parking. Plane. Hotel. Really, it is a pretty sweet deal.
I like BWI, the Baltimore Airport. It was very manageable, not overwhelming at all. I flew Delta out of BWI to Atlanta. A less manageable airport. There are seven (?) concourses. They are separated by train. I came in at one concourse and had to take a train to my connecting flight. It's a cool concept, but not exactly useful or user-friendly. What I did really like about ATL was there trash system. They have these special trash cans. They compact the trash every few hours, and apparently send a signal to staff when they need to be emptied. The trash is transported of site to a sorting facility. It is then sorted for recycling. Sometimes, it is plainly apparent that we live in the future. Of note, BWI and ATL have the same bathroom system. Exactly the same hardware for stalls, toilets, and automated faucets with automated soap dispensers. Kind of weird, like going to a mall in a different state and realizing it has the same architecture as the mall at home.
It turns out that I was lucky on this flight. I am next to the single empty seat on the flight! It seams that the seat is lacking a latch for the seat belt, so it can't be booked. Hooray for spreading out.
On a related note! Delta has started to charge $25 per check-in bag. This, by itself, is not that odd. They have however made the frame for the carry-on smaller! This means that the bag you thought would be fine because you have used it as a carry-on before (mine) was subject to a $25 charge. It seems as though I was prepared though. I had a daypack in my bag; I shifted my luggage around, and had a perfectly good carry-on. But, Delta, you have done something really stupid! By charging a ridiculous fee for checked luggage, you then encourage people to over-pack their carry-ons. What does this mean? There is no space in the cabin for luggage. Every overhead bin is packed, most people with something at their feet. Somehow, I don't think this is particularly good for business...
I know that by stating this, I might jinx it. There have been no crying babies. Really a much more pleasant experience that way.
So, I am going to this conference. I am supposed to table (advertise to represent my organization) for a few hours, participate in a discussion with all the other peace groups, but otherwise enjoy the conference. It is a goal of mine to at least once be considered important enough that someone pays for my transportation first-class. But this is cool too.
UPDATE: I wrote both of these posts on the plane. On Thursday. Delta naturally lost my bag. I just got it. And it had my compy's power cord in it. And I have so much to add about the conference, but I think sleep is probably the best option at the moment. Hopefully I will have a moment (or seventeen) to write about the conference tomorrow, else I certainly will on the plane coming back.
And so it begins...
It was surprisingly easy to unpack my car. Aside from the futon, it was only four trips of bringing things inside. I started to sort things in my room, allotting corners for this or that. Eventually, JK came up and asked if it was yet futon time (I had told him previously that I would need his help for that). I said that I had unpacked everything else, and that if he was available then now was a good time.
It took at least 20 minutes for AK and I to stuff the queen-size futon into my tiny sedan. And then, of course, it fills the space it occupies. I thought it would be quite awful trying to remove it. JK came along. He gave two tugs, and it was totally out. Wow. We brought it up to my room (second floor). He told me that he actually had a spare futon frame (and futon) that might go well underneath it. He fetched them, and again I was wowed at the ease with which he moved them. We discussed placement, and he help me hoist my futon (queen-size) atop his futon (full) atop the frame (somewhere between full and queen-size). He then disappeared into the wild unknown and I was left to make up my room.
I decided for that for the sake of the space I had, I would put fiction books by the bed, and non-fiction by the closet (Don't worry, I will post pictures). I also tried to decide what furniture was useful/important to try to fill the space.
I went to the nearby Walmart (Dad would love this: It is less than five minutes away!). It is huge. I told AK later that it could easily eat the Walmart by Hampshire. Twice. There is a full grocery section among other things. They also have cheaper furniture options than what I could find used (craigslist) or rented (yes, somehow Walmart's prices were about the same as renting furniture for just one month!). I decided that the best options for me would be a cheap "student" desk (with bookshelves!), a "bookcase" to be used for clothing, and various modular boxes for miscellany. I also bought a trash-can and a CO detector as my room was lacking. I returned the next day to purchase a space-heater. My room was lacking in much heat as well.
I went to the local grocery store, called "Bloom." It is seriously less than a minute from my apartment. I only drove becuase it was on the way back from Walmart. It is very nice. And big. It seems they like big down here. They have these touchscreens where you are able to search for products and it gives you an interactive map and is altogether very cool. I bought some staples, you know, like a six-pack of mac and cheese.
The people are so kind. It is like the sales associates actually want me to have a nice day. Very different from the Northeast. There is a touch of hick-ness too. But is so far more endearing than upsetting or anything else. And I love the accent. It really seems as though even a stranger genuinely cares. So different from what I am used to.
It is February 23, and still DM says that he thinks that I will probably start on March 1. Nothing more. Oh! I never mentioned the interesting turn! Towards the beginning of February, I called DM (as it seems that I always need to be the one to initiate, he only returns calls or emails, and is much better with calls), to try and get more details and he said, "Yea, my secretary reminded me of a way where you could have the same start data [March 1 at this point] and get paid for the work you do for us. You just have to go online and fill out this application. It is for the Summer Internship Program, but it doesn't have to be in summer." As it turns out, I had already applied for this! I quickly told him that I had submitted an application, and his secretary supposedly checked to make sure everything is in order. Apparently, he had to ask for special permission for me to start near immediately, blah blah blah. If it works, I will be paid at a rate of $2200 a month. No complaints here. But, it would be nice to actually have a firm start date!
Tuesday, February 23, 2010
The story so far
A while ago (probably over the summer), I decided that I would not be on
campus for the Spring semester. This could mean study abroad, exchange,
or field study. I had been considering Semester at Sea, but decided it
was not for me. I looked at a number of study abroad sites, and none of
them really clicked. I started investigating internship opportunities
domestically (that's the field study option).
campus for the Spring semester. This could mean study abroad, exchange,
or field study. I had been considering Semester at Sea, but decided it
was not for me. I looked at a number of study abroad sites, and none of
them really clicked. I started investigating internship opportunities
domestically (that's the field study option).
I asked CJ and CR if they knew of any labs or related that would not
mind a student intern. I was basically offering myself as free labor
(OK, so they had to pay me in experience/knowledge, but otherwise free)
in the hopes that I could find some sort of experiential learning
opportunity. CJ asked me to continue to pester him, to make sure he
would give me some information. He gave me three names of friends of
his with labs at the National Institutes of Health (NIH). I sent them
all cover letters and resumes, and the one whom CJ had thought least
likely emailed back to say, "Yes, I think we can arrange an internship.
I will get back to you next week. Give my best to CJ." This of course
is DM. My request was on 10/9/2010 (non-European dating, as in
October). The response was on 10/30. This should have set a precedent
in my mind. 20 days to respond.
Now, I was particularly excited about the prospect of working in a lab.
I had other choices of things to do for the Spring, but I decided that
working at the National Cancer Institute under DM would be really cool.
As I have said since the start of my Division II (major), "How can one
learn to communicate science, without knowing the science." I was going
to get a chance to know the science first-hand.
I paid attention to all of the deadlines for school. I filled out all
of my paperwork. I had to get JM's signature as he is my chair. He
basically thinks that I am off and doing things and learning, etc. so he
does not much care what he is signing for me. He is very happy to help
out in a bureaucratic way, but he also does not think we have to meet so
often if I am just going to ask him to sign things.
Hampshire has a January term. You take one class for three weeks in
January, meeting everyday. It is about equivalent to taking a class
over a semester (except that it is physically impossible to do the same
amount of reading in one month as a person might in four). I decided
that if I do not get this internship with DM, I would take "Gene
Cloning" over January term. I would get extensive "wet" lab experience
doing interesting molecular things. I asked CR if he thought that I
should take Gene Cloning (he teaches it) even if I end up with this
internship. He tried his best to talk me out of it without explicitly
saying, "Don't do it." Fine. I taught myself Cell Biology instead
(Alright, I am still continuing to teach myself Cell Biology almost two
months later).
Every few weeks, I would email DM and ask for more details. It was as
though he was enjoying tantalizing me; releasing just the tiniest morsel
of information at a time. He would say things like, "I have a meeting
on Thursday to work out details. I'll let you know." And then, he
wouldn't. Also, he never responded in less than five business days. It
became the end of the Fall semester (beginning of December), and I still
did not know if he wanted me there at the beginning of January. I had
already told school that they could have my room for the Spring, but I
did not yet know whether I would be there for January. I asked CJ what
chapters I should try to learn for Cell Biology. He told me to do most
of the non-molecular stuff. I wrote down which chapters he suggested.
DM said that I would start on Feb 1, but that he is still trying to work
it out. That's a relief. Parents can stop bugging me. I can look for
an apartment. I can even commit to scheduling things for January!
January was awesome. In addition to Cell Bio, I tried to learn some
linguistics as well, but that will have to be a whole different blog. I
really enjoyed the
learning-at-your-own-pace-with-no-external-responsibilities things. It
was pretty pleasant. I did have a job for Fall and theoretically
January (teaching Religious School at a local synagogue). But the
kids/parents could not get their acts together. So really I made my own
schedule and did what I wanted when I wanted. I even learned some Cell Bio.
I used roommates.com and craigslist to try and find potential
roommates/landlords. I found a few options, and in the middle of
January I went down to meet them, tour apartments, etc. I liked JK the
most, and I liked his condo the most. Not difficult. I also met with
DM for the first time to get to know him, ask questions in person
(rather than waiting for five days for a response), and actually hear
what the project was about. I barely understood what he was saying (if
I had not been studying Cell Bio, I really would not have understood).
Basically, all that molecular stuff that CJ said I would not need, I
needed. Badly.
I debriefed with CJ. He told me stories from the old days about DM. In
grad school, apparently DM had two girlfriends. One who knew about
both, one who didn't. It was a really nice conversation. I also had a
really nice chat with CR. It was the first time that I was really
chatting with him outside the bounds of the teacher-student setup. I
know that at Hampshire students are supposed to be like peers with the
faculty. That's why we call everyone by first name. Blah, blah, blah.
It is really hard to do that in practice. I am sorry to my faculty
members that may have felt otherwise. For me however, it is only now
(speaking for the end of January, beginning of February) that I am able
to appreciate faculty as peers. My bad. Sorry.
Somewhere in there, DM said that it would be the middle of February.
Then somewhere in there, he said the end of February or beginning of
March. Grr.
I took things into my own hands. I told JK that I would be moving in on
2/22. It worked. I did that.
Thanks for bearing with me. I should really write as it happens.
Unfortunately this blog was a late idea in the whole process. I promise
shorter posts in the future.
Subscribe to:
Posts (Atom)